Irei falar de duas doenças que são erros inatos do metabolismo, ou seja, deficiência na produção de uma proteína que nestes casos são de uma enzima e na PKU é a deficiência ou produção residual de até 5% da fenilalanina hidroxilase (PAH) e é esse tipo de mutação que classifica a doença:
Variantes da PKU
-PKU clássica: Crianças toleram 250-350mg phe/dia.
-PKU moderada: 350-400mg phe/dia.
-PKU branda: 400-600mg phe/dia para manter 300 mmol/L
-PKU atípica: erro no co-fator da PAH, a tetrahidrobiopterina BH4, co-fator essencial na
hidroxilação da Phe, aumentando indiretamente os níveis de Phe sanguínea. A BH4 é também requerida pela  tirosina hidroxilase e triptofano hidroxilase, enzimas que catalisam reações que precedem a síntese de neurotransmissores.
tirosina hidroxilase e triptofano hidroxilase, enzimas que catalisam reações que precedem a síntese de neurotransmissores.
-Kuvan: sintético da BH4 que aumenta a atividade da enzima.
Sintomas:
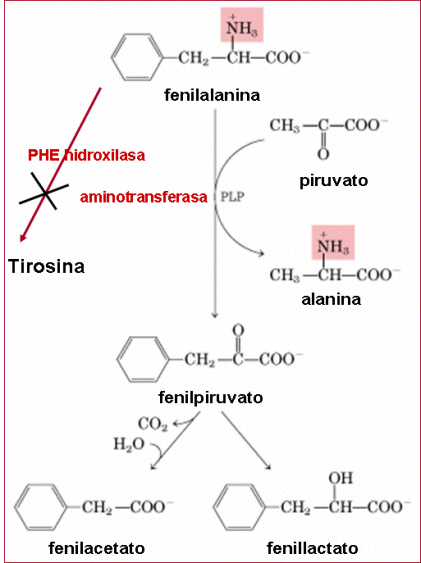
- a tirosina aminoácido não-essencial passa a ser pela falta de hidroxilação da phe, a tirosina hidroxilase pode catalisar a reação de phe em tirosina
-e por causa dos metabolitos formados apartir de uma via alternativa de transaminação de phe em fenilpiruvato, fenilacetato e fenilactato que inibem a piruvatokinase no cérebro e não no fígado é a causa do defeito de formação da mielina e a causa do retardo mental nesta doença, sintoma mais severo,
- o nome da doença vem do cheiro forte da urina pelo fenilacetato .
- Além deste sintoma existem também deficiência na pigmentação (cabelos e pele claros) devido à inibição completa da hidroxilação da tirosina pela tirosinase (primeira etapa na formação do pigmento melanina), complicações neurológicas e ocasionalmente atividade autística (autista), bem como transtornos de conduta, também podem se fazer presentes.
-A própria phe é uma das causas da complicação neurológica pois pode competir com outros aminoácidos pelo transporte através da barreira hematoencefálica resultando na falta de metabolitos essências desses aminoácidos no cérebro.
Tratamento:

Nenhum comentário:
Postar um comentário